 |
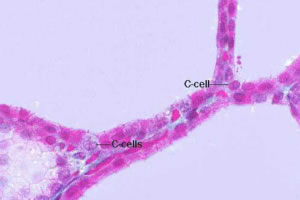
Il Carcinoma Midollare della Tiroide (CMT) deriva dalle cellule parafollicolari C della
tiroide, che fanno parte del sistema APUD.
Il CMT rappresenta il 5% circa di tutti i
carcinomi tiroidei con una frequenza di distribuzione uguale nei due sessi.
Metastatizza ai linfonodi paratracheali e cervicali, può diffondere per via ematogena al fegato, al polmone e alle ossa negli stadi avanzati della malattia. |
Durante l’embriogenesi, le cellule C migrano dalla cresta neurale all’interno della ghiandola tiroidea distribuendosi in maniera ubiquitaria, ma con una più alta concentrazione alla giunzione tra il 3° superiore e medio di ciascun lobo.
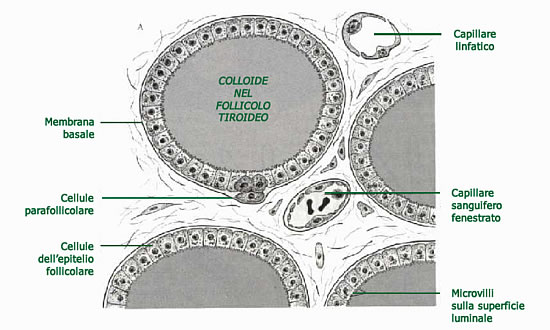
Si distinguono dalle cellule follicolari tiroidee per la differente capacità secretoria.
Le cellule C secernono infatti la calcitonina, un piccolo ormone peptidico di 32 aminoacidi, a catena singola, il cui gene è localizzato nel braccio corto del cromosoma 11.
Esistono varie forme di calcitonina ma, comunque, la concentrazione ematica non supera, in condizioni di normalità, i 10 pg/ml.
Il cut-off della calcitonina basale riportato in letteratura è variabile in rapporto ai diversi metodi di dosaggio. Secondo alcuni autori il limite di normalità è inferiore a 15-20 pg/ml.
Secondo alcuni autori valori compresi tra 20 e 100 pg/ml sono una “grey zone” da interpretare meglio con un test di stimolo con penta gastrina.
Non sempre modeste elevazioni della calcitonina sono indice di neoplasia midollare della tiroide.
Esistono infatti condizioni fisiologiche come la gravidanza, l’esercizio fisico strenuo, l’abuso di alcool o l’uso prolungato di alte dosi di PPI in cui la calcitonina ematica può essere lievemente aumentata.
L’uso continuato di inibitori di pompa protonica (PPI) per la cronica inibizione della secrezione acida gastrica potrebbe determinare per feed back una elevazione della gastrinemia che come saputoè un potente stimolo per la secrezione di calcitonina.
Una lieve ipercalcitoninemia può anche riscontrarsi in corso di insufficienza renale cronica, di tiroidite cronica autoimmune (con una prevalenza del 20%) ed anche nel 10-15% dei soggetti con tumori di origine neuroendocrina.
Per differenziare le ipercalcitoninemie neoplastiche e/o le recidive metastatiche di un CMT operato con quelle non neoplastiche viene usato (dove è possibile reperire le fiale di PEPTAVLON) il test di stimolo con pentagastrina.
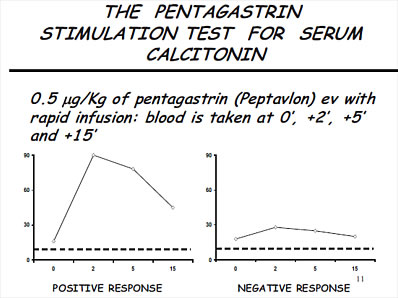
Viene iniettata endovena, molto rapidamente (in 5 secondi), una dose di pentagastrina pari a 0,5 mcg/Kg di peso corporeo, determinando la concentrazione di calcitoninemia basale, a 2, 5 e 7 minuti.
Una elevazione al picco, tra 2 e 5 minuti maggiore di 100 pg/ml o comunque di 3-5 volte il valore basale, è indicativa di una forma neoplastica. Concentrazioni inferiori a 30 o una curva piatta escluderebbero il CMT.
Vi sono anche pazienti con semplice iperplasia delle cellule C o con micro carcinoma midollare tiroideo con calcitoninemia basale di poco aumentata ma che al test di stimolo con peptavlon presentano una significativa impennata della concentrazione ematica.
Gli effetti collaterali predominanti del test di stimolo con penta gastrina sono rappresentati da nausea e vomito, precordialgia, tachicardia, sensazione di sapore metallico, crampi addominali ed nausea esofagei.
Un test di stimolo alternativo può essere effettuato con calcio ev con infusione in un minuto di 2mg/Kg
di peso corporeo e determinazione della calcitoninemia basale, a 1, 3, 5 e 10 minuti.
Valori di calcitonina al picco maggiori di 100 pg/ml o comunque di 3-5 volte il valore basale sono
indicativi di CMT.
Dal punto di vista epidemiologico esistono due forme di carcinoma midollare della tiroide: la forma sporadica (80%) e la forma familiare (20%) con differenti risvolti diagnostico terapeutici.
La forma sporadica ha un picco d’incidenza alla 5^ e 6^ decade di età e, di solito, si presenta come un nodulo singolo, unilaterale, scintigraficamente freddo, con un’incidenza massima dopo i 40 anni, con metastasi linfonodali frequenti alla diagnosi, sindrome diarroica spesso presente e calcificazioni del nodulo tiroideo.
La forma familiare si manifesta più frequentemente nella 2^ e 3^ decade nell’ambito delle MEN 2B, comunque sempre prima dei 40 anni con manifestazioni anche in età pediatrica; la neoplasiaè multifocale e bilaterale e viene trasmessa con modalità autosomica dominante.
Il carcinoma midollare familiare isolato, invece, predilige la 4^/6^ decade di vita.
Il CMT familiare ha una trasmissione autosomica dominante con una elevata penetranza, maggiore del 90%, con variabile espressività.
Vi sono, inoltre, altri fattori genetici che condizionano il grado di penetranza del CMT, come l’omozigosi o l’eterozigosi delle mutazioni del gene RET ed anche la presenza di polimorfismi
genetici.
Il diverso periodo di comparsa del doppio evento mutazionale rende ragione dei due fenotipi di CMT familiare, ad insorgenza precoce, e sporadico, ad insorgenza più tardiva. Inoltre il CMT familiareè multicentrico.
Da tenere presente inoltre che l’iperplasia delle cellule C viene considerata come una lesione precancerosa e può comparire anche in maniera sincrona con il CMT o precederne la comparsa.
L’etiopatogenesi del CMT sarebbe quella prevista dal modello di carcinogenesi ideato da Knudson “two hits” (a due colpi). Secondo tale noto modello cancerogenetico, nel CMT familiare, la prima tappa (first hit) consiste in una mutazione germinale che provoca l’iperplasia delle cellule C
parafollicolari. Il second hit è dato invece da una successiva mutazione somatica delle cellule C già “attivate”, provocando lo sviluppo del carcinoma midollare tiroideo.
Nel CMT sporadico il first e second hits sarebbero rappresentate da due successive e indipendenti mutazioni di tipo somatico.
|
Il principale gene associato al CMT è il proto-oncogene RET, di 55 Kilobasi, localizzato sul cromosoma 10q11.2 ed è espresso nelle
cellule derivanti dalla cresta neurale: cellule C
parafollicolari della tiroide, cellule cromaffini
della midollare surrenalica e cellule delle
ghiandole paratiroidee.
Esistono tre isoforme del gene RET (derivanti dallo splicing alternativo):
|
Il RET è composto da 21 esoni e codifica per un recettore transmembranario con attività tirosinochinasica. Mutazioni germinali attivanti del RET si riscontrano infatti nel 98% dei pazienti affetti da MEN 2A, nel 95% dei pazienti con MEN 2B e in circa l’88% dei soggetti affetti da FMTC.
Le mutazioni attivanti di tale protoncogene RET determinano l’autofosforilazione del recettore che diventa pertanto cronicamente attivato, indipendentemente dal legame con il ligando.
Ne consegue alterazione della crescita cellulare e carcinogenesi con correlazione tra le mutazioni del gene RET e il fenotipo dei pazienti affetti da CMT.
Tale correlazione tra mutazione genetica RET e fenotipo che ne deriva riveste importanza clinicopratica
tant’è che è stata proposta la tiroidectomia profilattica in presenza della sola mutazione germinale RET.
Lo screening genetico richiede solo un prelievo di sangue per l’estrazione del DNA linfocitario.
La diagnosi differenziale tra le diverse forme di CMT è importante sia per il trattamento che per il follow-up del paziente, sia per le scelte terapeutiche da seguire.
La differenziazione tra la forma sporadica e quella familiare si basa sull’analisi genetica RET.
In caso di positività della mutazione germinale RET è necessario ed obbligatorio escludere la copresenza di feocromocitoma e iperparatiroidismo con il dosaggio delle catecolamine plasmatiche ed urinarie, il dosaggio del PTH, calcemia, fosforemia, calciuria e fosfaturia 24 h per differenziare la MEN 2°A, la MEN 2B (questa con habitus caratterizzato dai neurinomi mucosi caratteristici, età precoce d’insorgenza e aggressività elevata) e il FMTC.
La terapia di elezione del CMT sia familiare che sporadico è la tiroidectomia totale di principio con svuotamento dei linfonodi del comparto centrale. Secondo alcuni autori, lo svuotamento e la dissezione degli altri compartimenti linfonodali andrebbe fatta solo in presenza di coinvolgimento clinico-ecografico; secondo altri andrebbe fatta l’asportazione bilaterale dei linfonodi centrali e laterali del collo. In ogni caso andrebbe fatta sempre l’esplorazione dei linfonodi laterali del collo e del mediastino superiore. Altri autori ancora ritengono attuabile l’asportazione del solo comparto linfonodale centrale e l.c ipsilaterali alla lesione, risparmiando quelli contro laterali, in particolare , nel CMT di diam max < di 1 cm. Alcuni chirurghi ritengono che si possa evitare la dissezione dei linfonodi cervicali controlaterali nel CMT unilaterale senza coinvolgimento dei linfonodi del comparto centrale e ipsilaterali l.c., indipendentemente dalla dimensione della neoplasia midollare.
Nel CMT familiare con malattia ancora non manifesta clinicamente, la tiroidectomia + svuotamento comparto centrale è tanto più risolutiva quanto più precocemente effettuata.
E’ dibattuto se effettuare di routine la dissezione linfonodale centrale nei pazienti giovani con mutazione RET ma senza manifestazioni cliniche e biochimiche, cioè con calcitonina normale sia basale che dopo stimolo con penta gastrina.
In presenza invece di metastasi linfonodali accertate, l’intervento è più demolitivo e comporta la dissezione della muscolatura e delle strutture centro-laterali del collo, con preservazione dello sternocleido-mastoideo.
In alcuni pazienti ancora potrebbe rendersi necessaria la dissezione mediastinicaa trans sternale, indicata comunque in tutti i CMT in stadio avanzato pT4.
La “reazione desmoplastica stromale” è un marcatore intraoperatorio di metastasi linfonodali.
La presenza di un denso stroma fibrotico con aumento di collagene che circonda le cellule neoplastiche e che non è presente nel tessuto ghiandolare tiroideo normale, sarebbe indice di possibili metastasi linfonodali con una sensibilità del 36% e una specificità prossima al 100%.
Prima dell’intervento di tiroidectomia nel CMT va sempre esclusa la presenza di feocromocitoma
per il noto rischio anestesiologico che può comportare.
Le percentuali di successo dei vari tipi di intervento sono variabili.
Comunque la percentuale dei soggetti che dopo l’intervento manifestano persistenza o ricorrenza
di malattia varia tra il 29 e l’85%.
Nei casi di MEN, la eventuale surrenalectomia, in presenza di feocromocitoma, dovrebbe essere effettuata prima della tiroidectomia.
Di recente, inoltre, si è affermato l’orientamento della “chirurgia profilattica codone-orientata” che si basa sulla forte e differenziata correlazione tra genotipo-fenotipo del CMT. Esiste infatti un algoritmo, dell’Università di Halle, che orienta il timing della tiroidectomia a seconda dei tre livelli differenti di rischio sulla base della tipologia di mutazione germinale presente e sulla base anche della risposta
della calcitonina al test di stimolo con penta gastrina.
Durante l’intervento il chirurgo deve localizzare ed esaminare le ghiandole paratiroidee. In caso di iperplasia o adenomatosi paratiroidea deve asportarle. Nel caso siano normali tenterà di preservarle o, in caso non sia possibile, può procedere al trapianto di un frammento ghiandolare nei muscoli del collo o nell’avambraccio.
Il follow.up dopo la tiroidectomia prevede l’instaurazione di terapia sostitutiva con L-T4 con calibrazione posologica a 30 e 60 giorni dall’intervento, mantenendo il TSH nel range normale basso della norma (magari intorno a 0,50 mcU/ml).
La determinazione della calcitonina deve essere effettuata a 30 e 60 giorni dalla tiroidectomia.
Se i valori basali di calcitonina risultano indosabili dovrebbe essere effettuato un test di stimolo con penta gastrina per confermare la radicalità dell’intervento e l’assenza di MTS.
I soggetti che presentano dopo l’intervento calcitonina indosabile e test di stimolo negativo in due distinte occasioni possono considerarsi clinicamente guariti.
In genere comunque la sopravvivenza del CMT a 10 anni è di circa il 60-70%.
Inoltre,anche nei soggetti in cui la neoplasia è scoperta precocemente con i test genetici la remissione clinica è completa.
Il dosaggio del CEA (antigene carcinoembrionario) è un esame di secondo livello, perché spesso risulta normale in soggetti con CTM occulto. Secondo alcuni autori il CEA è utile nell’individuazione
dei pazienti ad alto rischio, con coinvolgimento linfonodale e neoplasia midollare progressiva.
Anche la cromogranina A correlerebbe in maniera diretta con i livelli di calcitoninemia.
Un po’ in disuso, come ausilio diagnostico, il dosaggio della NSE (Enolasi Neurono
Specifica), che può trovarsi elevata in corso di CMT.
In caso di calcitonina dosabile o test di stimolo positivo dopo l’intervento bisogna ricercare
eventuale recidiva o localizzazione MTS secondaria.
La tecniche di imaging di 1° livello per localizzare eventuali MTS comprendono: la TAC e la RMN
di collo, torace e addome, l’ecografia del collo e dell’addome e la scintigrafia ossea.
Tuttavia la localizzazione delle ricorrenze tumorali del CMT può essere particolarmente difficile, soprattutto nei casi di calcitoninemia solo lievemente aumentata. In tali casi si può ricorrere al cateterismo selettivo con determinazione del gradiente per la calcitonina, alla laparoscopia o a tecniche di imaging nucleare con impiego di radionuclidi particolari: I131MIBG, T99-DMSA, Tl201, I131antiCEA, I131AntiCalcitonina, Tc99mSestamibi e la PET.
La terapia del carcinoma midollare tiroideo metastatico può basarsi sulla radioterapia di ev lesioni MTS ossee e della chemioterapia. La chemioterapia si basa sull’uso della doxorubicina, del cisplatino o sulla polichemioterapia combinata ciclofosfamide-vincristina-dacarbazina o sull’associazione dacarbazina- 5- fluorouracile.
Vandetanib (CAPRELSA da 300 mg cpr) e cabozantinib (COMETRIQ 140 mg die: 1 cp arancione da 80 mg + 3 cp grigie da 20 mg die), inibitori di protein-kinasi sono invece i nuovi farmaci efficacissimi e di recente commercializzati e approvati nella terapia dei carcinomi midollari, nelle forme avanzate della malattia.
Nel caso di neoplasia residua intraparenchimale tiroidea, di qualche utilità può essere il radioiodio
per l’effetto indiretto sulle cellule C parafollicolari, essendo captato dalla cellule follicolari tiroidee
adiacenti.
Vi sono studi sulla terapia cosiddetta pRAIT (pretargeted radioimmunotherapy) con impiego di apteni bivalenti marcati con I131 associati ad anticorpi monoclonali. Tuttavia gli effetti collaterali di epato e mielotossicità non sono indifferenti.
Comunque, i trattamenti chemioterapici, nei diversi schemi utilizzati, riducono la progressione del CMT per un periodo compreso tra 4 e 29 mesi con sopravvivenze variabili da 8 a 33 mesi n ei pazientiresponders alla chemio e tra 3 e 20 mesi nei non responders al trattamento.
In fase di sperimentazione farmaci promettenti che inibiscono sia la neoangiogenesi tumorale che il gene RET mutato nel CMT. Gli Oncologi del Fred Hutchinson Research Center di Seattle hanno sperimentato il sunitinib che, oltre a bloccare la formazione di nuovi vasi sanguigni, agirebbe anche inibendo il gene RET. Altre molecole promettenti sono il pazopanib, sperimentato alla Mayo Clinic di Rochester, anch’esso con azione antiangiogenetica e inibente il RET e l’XL 184-301, che agisce come inibitore della Ret-tirosinchinasi e di altri fattori di crescita promotori dell’angiogenesi,
quest’ultimo in sperimentazione nel centro fiorentino.
Allo stato attuale gli unici farmaci approvati ed utilizzabili con efficacia dimostrata sono,
come già detto il Vandetanib e cabozantinib.
RIFERIMENTI BIBLIOGRAFICI:
- Malattie della Tiroide di Fabrizio Monaco, Soc.ed.Universo Cap 5D Ed 2007
- Endocrinologia oggi: Carcinoma Midollare della Tiroide di Furio Pacini
- Diagnostica tiroidea: un update – follow-up del cancro della tiroide di Roberto
Castello. Società italiana medicina di laboratorio – Rimini 30 Ottobre 2008
- Carcinoma Midollare della tiroide e screening genetico di Rocchetti, Braccioli et al Clinica endocrinol. Univ. Di Ancona. Journal of Medicine 2004
- Diagnosi genetica del carcinoma midollare della tiroide: implicazioni diagnostiche e terapeutiche. L’Endocrinologo 2004 (Elisei-Pinchera et al.)
- Prontuario di terapia endocrina e metabolica di F.monaco, Giuliani Ed SEU 2006
- Guyetant et al: C-Cell hyperplasia associated with chronic lymphocytic thyroiditis Hum Pathol
1994.
- Lacka et al: Usefulness of serum calcitonin, CEA and AFP assays in the early detection of medullary thyroid carcinoma relapse. Wiad Lek 2002.
- Szinnai et al: Review of multiple endocrine neaoplasia type 2A in children: therapeutic results of early thyroidectomy and prognostic value of codon analysis. Pediatrics 2003
- Wells et al: Predictive DNA testing and prophylactic thyroidectomy in patients at risk for multiple endocrine neoplasia type 2A. Ann Surg,194,220,3237.
- Robinson et al: Medullary thyroid carcinoma, in Endocrinology, V edition- 2006
|








{kind=link}